
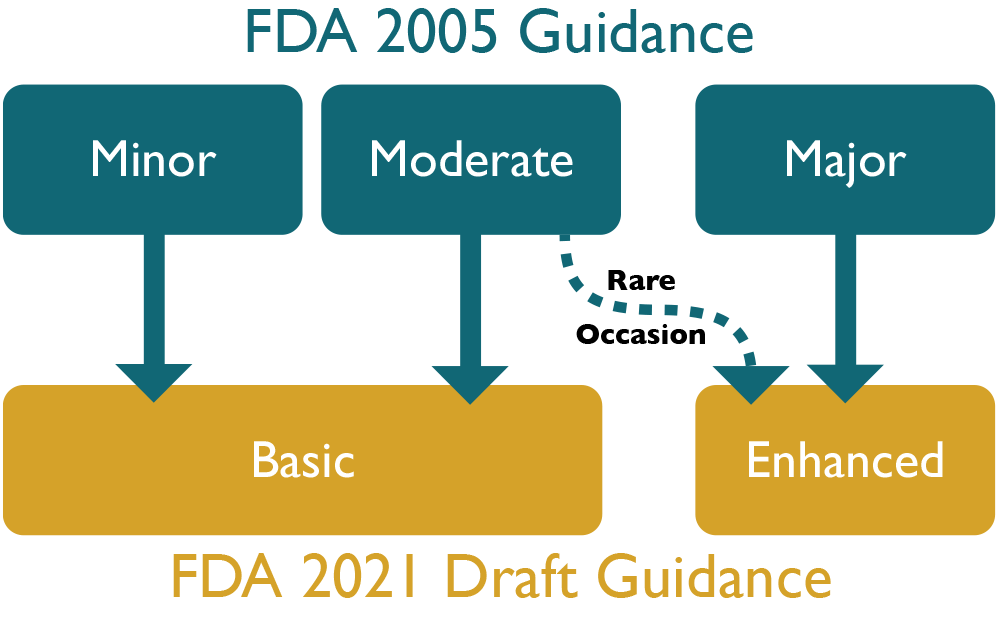
Episode 2: Deciphering New FDA Guidance on Software Submissions
- Posted by Jimmy Townsend and Nathan Viehmann
- On March 24, 2022

ABOUT THE AUTHOR(S)

Jimmy Townsend
Jimmy is an embedded software expert, with nearly two decades of experience. He is a primary contributor to 62304 based software and excels at quality procedures with an emphasis on creating outputs that are compliant in a minimally burdensome way. He has broad technical exposure from bare-metal hardware to Android 8, rapid development consumer devices to class III medical devices, and everything in between.

Nathan Viehmann
Nathan is an experienced software engineer; bringing well-crafted software to a variety of medical devices and high technology systems. His role at Engenious often means taking science fiction and bringing it to reality utilizing his skills in multiple programming languages, PCB Board Design and Software Management.

